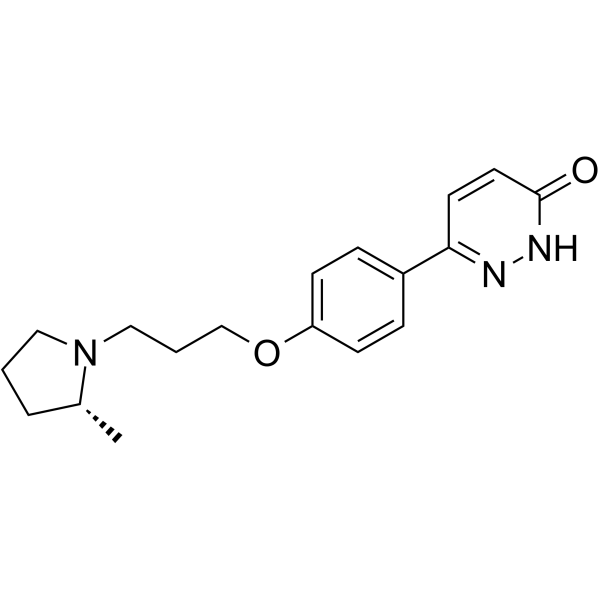
Irdabisant (CEP-26401) 是一种具有选择性、口服活性和血脑屏障渗透性的组胺 H3 受体 (histamine H3 receptor, H3R) 拮抗剂/逆激动剂,对大鼠 H3R 和人 H3R 的Ki分别为 7.2 nM 和 2.0 nM。Irdabisant 对 hERG 电流的抑制活性相对较低,IC50为 13.8 μM。Irdabisant 在大鼠社会认知模型中具有认知增强和唤醒作用。Irdabisant 可用于精神分裂症或认知障碍的研究。
| 生物活性 | Irdabisant (CEP-26401) is a selective, orally active and blood-brain barrier (BBB) penetranthistamine H3 receptor(H3R) inverse agonist/inverse agonist withKivalues of 7.2 nM and 2.0 nM for rat H3R and human H3R, respectively. Irdabisant has relatively low inhibitory activity against hERG current with an IC50of 13.8 μM. Irdabisant has cognition-enhancing and wake-promoting activities in the rat social recognition model. Irdabisant can be used to research schizophrenia or cognitive impairment[1][2]. |
| IC50& Target[1] | rat H3receptor 7.2 nM (Ki) | human H3receptor 2 nM (Ki) |
|
体外研究
(In Vitro) | Irdabisant (CEP-26401, compound 8a) shows antagonist activity with Kb, appvalues of 1.0 nM and 0.4 nM for rat H3R and human H3R, respectively; shows inverse agonist activity with EC50values of 2.0 nM and 1.1 nM for rat H3R and human H3R, respectively[1].
Irdabisant has moderate activity at Muscarinic M2(Ki= 3.7 ± 0.0 μM) and Adrenergic α1A(Ki= 9.8 ± 0.3 μM) receptors, Dopamine transporters (Ki= 11 ± 2 μM), Norepinephrine transporters (Ki= 10 ± 1 μM), and phosphodiesterase PDE3 (IC50= 15 ± 1 μM)[1].
Irdabisant inhibits the cytochrome P450 enzymes CYP1A2, 2C9, 2C19, 2D6, and 3A4 with IC50values of greater than 30 μM, indicating less potential for drug-drug interactions[1].
|
体内研究
(In Vivo) | CEP-26401 (0.01-0.3 mg/kg; p.o.; single dosage) dose-dependently inhibits H3R agonistRAMH-induced dipsogenia[1].
CEP-26401 (0.0001-0.1 mg/kg; i.v. or p.o.; single dosage) improves performance in the rat social recognition model of short-term memory[1].
CEP-26401 (3-30 mg/kg; p.o.; single dosage) exhibits wake-promoting activity in rat[2].
CEP-26401 (3-30 mg/kg; i.p.) increases prepulse inhibition (PPI) in DBA/2NCrl mice[2].
CEP-26401 (1 mg/kg for i.v. and 3 mg/kg for p.o.; single dosage) is rapidly absorbed with high oral bioavailability in rat and monkey, and shows a moderate clearance in monkey and dog compared to the rat[1].
Pharmacokinetic Parameters of Irdabisant (compound 8a) in rats, dogs and monkeys[1].
| Rat | Dog | Monkey | | i.v.t1/2(h) | 2.6 | 2.9 | 5.4 | | i.v.Vd(L/kg) | 9.4 | 3.5 ± 1.1 | 3.8 ± 0.9 | | i.v.CL (mL/min/kg) | 42 | 13.2 ± 1.5 | 7.7 ± 1.8 | | p.o.t1/2(L/kg) | 2.9 | 2.7 | 5.0 | | p.o.AUC (ng·h/mL) | 984 | 1190 ± 180 | 1919 ± 611 | | p.o.Cmax(ng/mL) | 270 | 230 ± 70 | 760 ± 74 | | p.o.F (%) | 83 | 22 ± 2 | 83 ± 18 | | Brain to plasma ratio | 2.6 ± 0.2 | 2.4 ± 0.4 | / |
| Animal Model: | Male Sprague-Dawley rats (i.p. 10 mg/kg RAMH-induced dipsogenia model)[1] | | Dosage: | 0.01-0.3 mg/kg | | Administration: | p.o.; single dosage | | Result: | Dose-dependently inhibited H3R agonistRAMH(HY-100999)-induced dipsogenia (which manifests as water drinking) with an EC50value of 0.06 mg/kg. |
| Animal Model: | Male Sprague-Dawley rats (adult rats were briefly exposed to a juvenile rat for build social recognition model)[2] | | Dosage: | 0.0001, 0.001, 0.01 and 0.1 mg/kg for i.p.; 0.01 and 0.1 mg/kg for p.o. | | Administration: | i.v. or p.o.; single dosage | | Result: | Effectively reduced the ratio of investigation duration (RID) at doses over the range from 0.001 to 0.1 mg/kg i.p. and at 0.01 and 0.1 mg/kg p.o., demonstrating potent enhancement of short-term sensory memory in this model. |
| Animal Model: | Male Sprague-Dawley rats[2] | | Dosage: | 3, 10 and 30 mg/kg | | Administration: | p.o.; single dosage | | Result: | Exhibited robust wake promotion with the treated animals awake 90% of the time up to 3 h postdosing at 30 mg/kg. |
| Animal Model: | Male DBA/2NCrl mice (19-27 g; 7-9 weeks)[2] | | Dosage: | 3, 10 and 30 mg/kg | | Administration: | i.p.; single dosage | | Result: | Increased prepulse inhibition (PPI) in DBA/2NCrl mice, whereas the antipsychoticRisperidone(HY-11018) is effective at 0.3 and 1 mg/kg i.p.. |
| Animal Model: | Male Sprague-Dawley rats, male beagle dogs and male cynomolgus monkeys[1] | | Dosage: | 1 mg/kg for i.v. and 3 mg/kg for p.o. | | Administration: | i.v. and p.o. | | Result: | Exhibited rapid absorption with high oral bioavailability in rat and monkey, and showed a moderate clearance in monkey and dog compared to the rat. |
|
| Clinical Trial | |
| 分子量 | |
| Formula | |
| CAS 号 | |
| 运输条件 | Room temperature in continental US; may vary elsewhere. |
| 储存方式 | Please store the product under the recommended conditions in the Certificate of Analysis. |
 m.cnreagent.com
m.cnreagent.com