生物活性
体外研究:MK-8617在体外不是细胞色素p450酶的抑制剂(IC50),CYP1A2,3A4,2B6,2C9,2C19或2D6,>60μM,是体外1.6μMCYP2C8的中度可逆抑制剂。 当与放射性配体结合和酶法测定的一般组合以10μM进行筛选时,MK-8617无活性。
体内研究:氚标记的MK-8617在大鼠,狗和猴(<10%turover)的肝微粒体(+ NADPH)中表现出最低的代谢周转率,60分钟后,人肝微粒体(34%转换)显着转换(10μM化合物,1mg / mL微粒体蛋白)。在药代动力学方面,MK-8617在各种物种(36-71%)之间表现出良好的口服生物利用度,具有较低的间隙和分布体积。该化合物在临床前物种中的消除半衰期相对较长。在小鼠(C57Bl / 6)中,单次剂量5和15mpk po(n = 3)导致在复合物攻击后3天和4天测量的循环网织红细胞增加。在大鼠(Sprague-Dawley)中,1.5,5和15mpk po(n = 5)的单次剂量滴定导致血清促红细胞生成素(EPO)水平相对于1.7倍,8倍和204倍的大幅增加车辆。在攻击后3天以5和15mg / kg观察循环网织红细胞的增加,并且在攻击后4天用15mg剂量观察。
化学数据
| 分子量 | 443.45 |
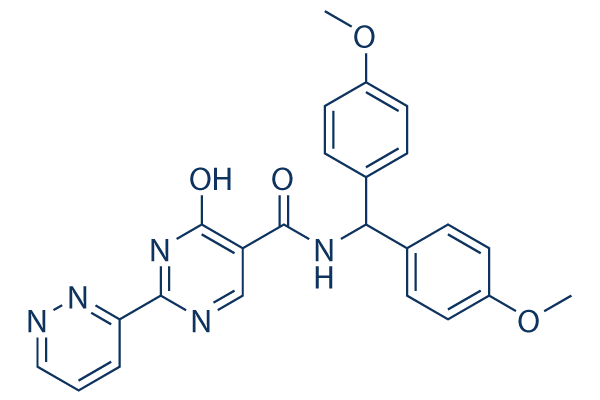
| 分子式 | C24H21N5O4 |
| CAS号 | 1187990-87-9 |
| 纯度 | >98% |
| 溶解性(25°C) | 88 mg/mL in DMSO |
| 储存和运输条件 | 固体粉末: -20°C 冷藏长期储存
常温运输及临时存放 |
实验操作 来自于公开的文献,仅供相同实验参考(如实验材料、目的不同,请参考其他文献)
| 动物实验 |
|---|
| 动物模型 | C57Bl/6 mice |
| 配制 | DMSO/PEG400/water (5:40:55, v/v/v) |
| 剂量 | 1 mg/kg po, 0.5 mg/kg iv |
| 给药处理 | i.v or p.o |
不同实验动物依据体表面积的等效剂量转换表(数据来源于FDA指南)
| 小鼠 | 大鼠 | 兔 | 豚鼠 | 仓鼠 | 狗 |
| 重量 (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| 体表面积 (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km系数 | 3 | 6 | 12 | 8 | 5 | 20 |
| 动物 A (mg/kg) = 动物 B (mg/kg) × | 动物 B的Km系数 |
| 动物 A的Km系数 |
例如,依据体表面积折算法,将化合物用于小鼠的剂量20 mg/kg 换算成大鼠的剂量,需要将20 mg/kg 乘以小鼠的Km系数(3),再除以大鼠的Km系数(6),得到化合物用于大鼠的等效剂量为10 mg/kg。
储备液配制
以下数据基于产品分子量,对于特殊产品,请参照COA中的储备液配制条件和说明进行操作。
| Concentration / Solvent Volume / Mass | 1 mg | 5 mg | 10 mg |
|---|
| 1 mM | 2.255 mL | 11.2752 mL | 22.5505 mL |
| 5 mM | 0.451 mL | 2.255 mL | 4.5101 mL |
| 10 mM | 0.2255 mL | 1.1275 mL | 2.255 mL |
 m.cnreagent.com
m.cnreagent.com