| 包装 | 价格(元) |
| 5mg | 电议 |
| 10mg | 电议 |
| 100mg | 电议 |
| 200mg | 电议 |
Kinase experiment: | The enzymatic kinase activity is assessed by measuring the phosphorylation of a synthetic substrate by the purified GST-fusion FGFR3-K650E kinase domain, in the presence of radiolabeled ATP. Enzyme activities are measured by mixing 10 μL of a 3-fold concentrated Infigratinib solution or control with 10 μL of the corresponding substrate mixture (peptidic substrate, ATP and [γ33P]ATP). The reactions are initiated by addition of 10 μL of a 3-fold concentrated solution of the enzyme in assay buffer. The final concentrations of the assay components are as following: 10 ng of GST-FGFR3-K650E, 20 mM Tris-HCl, pH 7.5, 3 mM MnCl2, 3 mM MgCl2, 1 mM DTT, 250 μg/mL PEG 20000, 2 μg/mL poly(EY) 4:1, 1% DMSO and 0.5 μM ATP (γ-[33P]-ATP 0.1 μCi). The assay is carried out according to the filter binding (FB) method in 96-well plates at room temperature for 10 min in a final volume of 30 μL including the components as indicated above. The enzymatic reactions are stopped by the addition of 20 μL of 125 mM EDTA, and the incorporation of 33P into the polypeptidic substrates is quantified as following: 30 μL of the stopped reaction mixture are transferred onto Immobilon-PVDF membranes previously soaked for 5 min with methanol, rinsed with water, soaked for 5 min with 0.5% H3PO4, and mounted on vacuum manifold with disconnected vacuum source. After spotting, vacuum is connected, and each well rinsed with 0.5% H3PO4 (200 μL). Free membranes are removed and washed four times on a shaker with 1% H3PO4 and once with ethanol. Membranes are dried and overlaid with addition of 10 μL/well of a scintillation fluid. The plates are eventually sealed and counted in a microplate scintillation counter. IC50 values are calculated by linear regression analysis of the percentage inhibition of NVP-BGJ398[1]. |
Cell experiment: | Murine BaF3 cell lines are cultured in RPMI-1640 media supplemented with 10% FBS, 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, and Pen/Strep. Cells are passaged twice weekly. Compound-mediated inhibition of BaF3 cell proliferation and viability is assessed using a Luciferase bioluminescent assay. Exponentially growing BaF3 or BaF3 Tel-TK cells are seeded into 384-well plates (4250 cells/well) at 50 μL/well using a μFill liquid dispenser in fresh medium. Infigratinib is serially diluted in DMSO and arrayed in a polypropylene 384-well plate. Then 50 nL of compound are transferred into the plates containing the cells by using the pintool transfer device, and the plates incubated at 37℃ (5% CO2) for 48 h. Then 25 μL of Bright-Glo are added, and luminescence is quantified using an Analyst-GT. Custom curve-fitting software is used to produce a logistic fit of percent cell viability as a function of the logarithm of inhibitor concentration. The IC50 value is determined as the concentration of compound needed to reduce cell viability to 50% of a DMSO control[1]. |
Animal experiment: | Mice[1] Female HsdNpa: Athymic Nude-nu mice are used. Infigratinib is formulated as a suspension in PEG300/D5W (2:1, v/v) and administered orally for 12 consecutive days at the doses of 10 and 30 mg/kg/qd. Tumor and body weight data are analyzed by ANOVA with post hoc Dunnett’s test for comparison of treatment versus control group. The post hoc Tukey test is used for intragroup comparison. Statistical analysis is performed using GraphPad prism 4.02. As a measure of efficacy, the T/C (%) value is calculated. Rats[1] Female nude Rowett rats 6-9 weeks of age are used. Infigratinib is formulated as a solution in acetic acid-acetate buffer pH 4.6/PEG300 (1:1, v/v) and applied daily by gavage to the tumor-bearing rats (n=8) for 20 consecutive days at doses of 5, 10, and 15 mg/kg/qd (free base equivalents). The application volume is 5 mL/kg. Tumor volumes are measured with calipers and determined according to the formula: length×width×height×π/6. Antitumor activity is expressed as T/C (%): (mean change of tumor volume of treated animals/mean change of tumor volume of control animals)×100. Regressions (%) are calculated. |
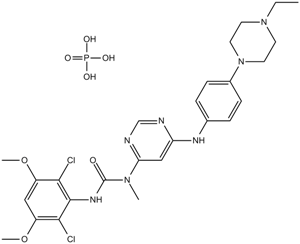
| 产品描述 | Description: IC50 Value: 0.9 nM (FGFR1); 1.4 nM (FGFR2); 1 nM (FGFR2) [1] NVP-BGJ398 is a novel selective, pan-specific FGFR inhibitor currently in Phase I clinical trials for cancer therapy. in vitro: NVP-BGJ398 is a novel and selective fibroblast growth factor receptor (FGFR) inhibitor. NVP-BGJ398 inhibit FGFR1, FGFR2, FGFR3 with IC50 of 0.9 nM, 1.4 nM and 1 nM. NVP-BGJ398 inhibited FGFR1, FGFR2, and FGFR3 with single digit nmol/L IC50 in biochemical and cellular autophosphorylation assays and FGFR4 with 38- to 60-fold lower potency. NVP-BGJ398 significantly inhibits proliferation of cancer cell lines bearing FGF/FGFR genetic alterations across various cancer types.Among the 35 cell lines selected from the high-throughput assays, 28 were confirmed as sensitive to NVP-BGJ398 with IC50s ranging from 0.001 to 500 nM. Cancer cell lines harboring FGF19 copy number gain at the 11q13 amplicon are sensitive to NVP-BGJ398 only when concomitant expression of beta-klotho occurs [1]. Cell lines with activating FGFR2 mutations (S252W, N550K) were more sensitive to dovitinib or NVP-BGJ398 when compared with their FGFR2 wild-type counterparts (P = 0.073 and P = 0.021, respectively). Both agents inhibited FGFR2 signaling, induced cell-cycle arrest, and significantly increased apoptosis in FGFR2-mutant lines. In vitro, dovitinib and NVP-BGJ398 were both potent at inhibiting cell growth of FGFR2-mutant endometrial cancer cells, but the activity of dovitinib was less restricted to FGFR2-mutant lines when compared with NVP-BGJ398 [2]. in vivo: In tumor tissues from primary tumor model GAM033 treated with 15 mg/kg NVP-BGJ398, NVP-BGJ398 shows inhibition to FGFR and ERK1/2 [1].NVP-BGJ398 significantly inhibited the growth of FGFR2-mutated endometrial cancer xenograft models [2]. Toxicity: N/A Clinical trial: Phase 1 |
 m.cnreagent.com
m.cnreagent.com