| CAS NO: | 1143532-39-1 |
| 包装 | 价格(元) |
| 5mg | 电议 |
| 25mg | 电议 |
| 100mg | 电议 |
| Physical Appearance | A solid |
| Storage | Store at -20°C |
| M.Wt | 428.92 |
| Cas No. | 1143532-39-1 |
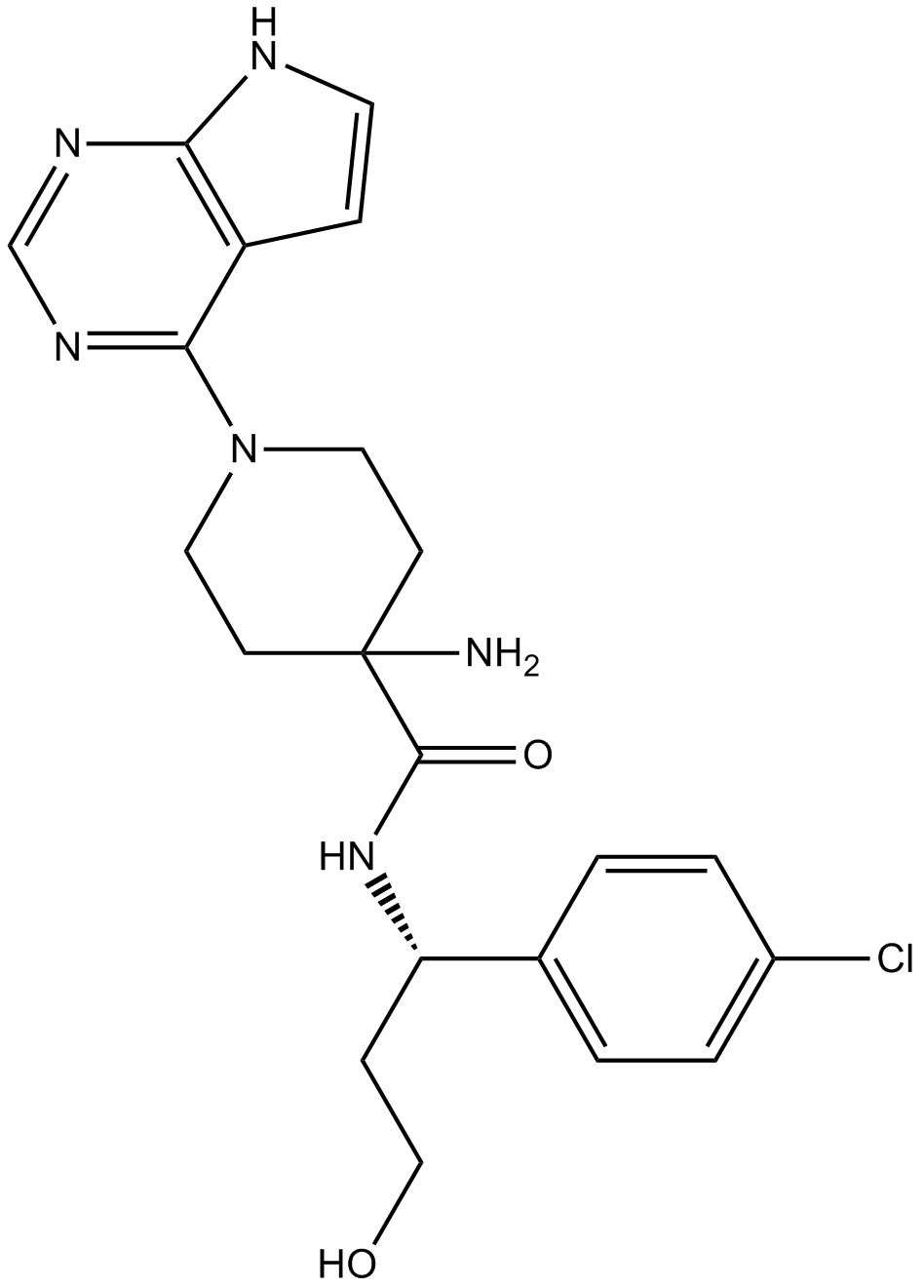
| Formula | C21H25ClN6O2 |
| Solubility | ≥21.45 mg/mL in DMSO; insoluble in H2O; ≥5.04 mg/mL in EtOH with ultrasonic |
| Chemical Name | 4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide |
| Canonical SMILES | C1CN(CCC1(C(=O)NC(CCO)C2=CC=C(C=C2)Cl)N)C3=NC=NC4=C3C=CN4 |
| 运输条件 | 蓝冰运输或根据您的需求运输。 |
| 一般建议 | 为了使其更好的溶解,请用37℃加热试管并在超声波水浴中震动片刻。不同厂家不同批次产品溶解度各有差异,仅做参考。若实验所需浓度过大至产品溶解极限,请添加助溶剂助溶或自行调整浓度。溶液形式一般不宜长期储存,请尽快用完。 |
AZD5363是一种新型有效的磷酸肌醇-3-激酶(PI3K)/ Akt信号通路抑制剂,IC50值约为200 nM[1]。
AZD5363抑制阉割性前列腺癌(CRPC)的发展。AZD5363诱导簇集蛋白(CLU)和自噬,可能作为细胞保护反应,可以影响PI3K/Akt下游信号[2]。AZD5363以剂量依赖的方式抑制许多人类肿瘤细胞的生长,该作用方式可以是单一疗法,也可以与HER2的抑制剂联合用于乳腺癌模型中[3]。切割PARP的表达及caspase 3的活性检测表明,AZD5363诱导细胞凋亡[1]。
最重要的是,AZD5363在体内显著靶向PI3K/Akt信号通路,从而减少血清PSA水平和肿瘤体积,最终,AZD5363可能推迟CRPC的发展[1]。
参考文献:
[1] Thomas C, Crafter C, Davies B, Zoubeidi A, Gleave ME. AZD5363, A novel AKT inhibitor, delays prostate cancer progression. The Journal of Urology. May 2011. 185(4S): e292-293.
[2] Kumano M, Zhang F, Shiota M, Crafter C, Davies B, Zoubeidi A, Gleave M. Clusterin knockdown enhances antitumor activity of a novel Akt inhibitor, AZD5363, through inhibition of autophagy in prostate cancer. The Journal of Urology. May 21 2012. e392.
[3] Davies BR, Greenwood H, Dudley P, et al. Preclinical Pharmacology of AZD5363, an Inhibitor of AKT: Pharmacodynamics, Antitumor Activity, and Correlation of Monotherapy Activity with Genetic Background. Mol. Cancer Ther. Arp 2012. 11: 873.
| 细胞实验[1] : | |
细胞系 | BT474c(Her2? PIK3CA突变乳腺),LNCaP(PTEN-nul前列腺)和MDA-MB-468(PTEN-nul乳腺)癌细胞中的GSK3 |
溶解方法 | 该化合物在DMSO中的溶解度大于10mM。若配制更高浓度的溶液,一般步骤如下:请将试管置于37℃加热10分钟和/或将其置于超声波浴中震荡一段时间。原液于-20℃可放置数月。 |
反应条件 | pGSK3β(IC50:BT474c中为0.76μM,LNCaP中为0.06μM,MDA-MB-468中为0.38μM)pPRAS40(IC50:BT474c中为0.31μM,LNCaP中为0.22μM,MDA-MB-468中为0.39μM)pFOXO3a染色体易位(IC50:BT474c中为0.69μM) |
实验结果 | AZD5363抑制AKT底物的磷酸化,在3个细胞系中的IC50值为0.06至0.76μM。AZD5363还有效抑制BT474c细胞和LNCaP细胞中S6和4E-BP1的磷酸化。 |
| 动物实验[1] : | |
动物模型 | BT474c肿瘤异种移植裸鼠 |
剂量 | 溶于DMSO/Kleptose缓冲液,口服给药300或100mg/kg急性剂量的AZD5363。 |
实验结果 | 裸鼠中,口服给药AZD5363以剂量和时间依赖性的方式减少PRAS40、GSK3和S6的磷酸化。给予300mg/kg剂量的AZD 5363后显著抑制3种生物标志物的磷酸化,抑制效果的持续时间至少24小时。100mg/kg剂量的AZD5363显著抑制3种生物标志物的磷酸化,抑制效果的持续时间至少8小时。 |
注意事项 | 请测试所有化合物在室内的溶解度,实际溶解度和理论值可能略有不同。这是由实验系统的误差引起的,属于正常现象。 |
References: [1] Davies B R, Greenwood H, Dudley P, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Molecular cancer therapeutics, 2012, 11(4): 873-887. | |
| 描述 | AZD5363是吡咯并嘧啶衍生的Akt抑制剂,IC50值小于10 nM。 | |||||
| 靶点 | Akt | |||||
| IC50 | <10 nM | |||||
 m.cnreagent.com
m.cnreagent.com