| 包装 | 价格(元) |
| 5mg | 电议 |
| 10mg | 电议 |
| 25mg | 电议 |
Cell experiment: | In vitro toxicity assays are performed on primary AML mononuclear cells over a 48 h period using a MTS cell proliferation assay. Lethal doses (LD50) are calculated. Cells are treated with voreloxin (31.25 nM to 4 μM) and Ara-C (62.5 nM to 8 μM) by serial dilution and incubated for 48 h in a final volume of 90 μL. Following the incubation, 20 μL of MTS reagent are added and the reaction is incubated for a further 4 h. The absorbance of the reaction after this time is read by spectrophotometry at 490 nm and the percentage of viable cells calculated relative to untreated control cells in the same assay. IC50 values are calculated using Calcusyn software[3]. |
Animal experiment: | Animals are weighed, randomized by body weight, and assigned to the study groups before initiation of treatment. Voreloxin is administered intravenously (IV) at 10 or 20 mg/kg once on day zero and once on day four (q4d ×2). Cytarabine is administered subcutaneously (SC) at 20 or 60 mg/kg every 8 h on day zero and day four (tid q4d ×2). Tissues and blood are sampled on days 6, 8, and 12 from at least three and not greater than ten animals per treatment group. Femurs are placed in Streck Tissue Fixative solution, or in 10% formalin solution for 24-48 h followed by a 70% dehydrant (ethanol, isopropanol, methanol). Femurs are decalcified, paraffin embedded, and sectioned at Biopathology Labs. The four micron sections are stained with hematoxylin-eosin (H&E). H&E stained femurs are examined and percent cellularity of the bone marrow is determined. Digital photographs of representative femur sections are taken on a Leica DM2000 microscope using Image-Pro Plus v6.1 software[2]. |
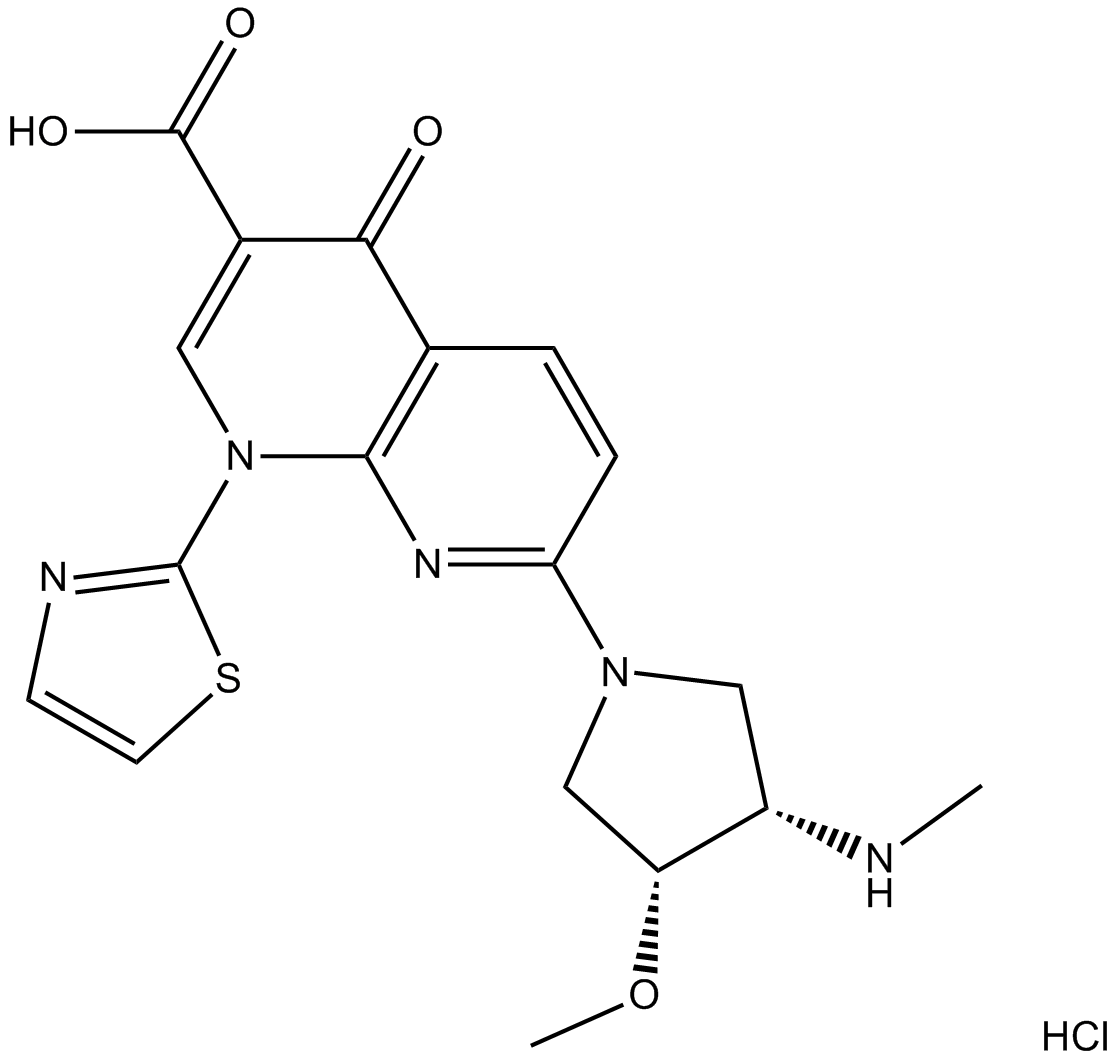
| 产品描述 | Voreloxin Hydrochloride is a first-in-class topoisomerase II inhibitor that intercalates DNA and induces site-selective DNA DSB, G2 arrest, and apoptosis. Voreloxin Hydrochloride is a first-in-class topoisomerase II poison and inhibitor that intercalates DNA and induces site-selective DNA DSB, G2 arrest, and apoptosis. Voreloxin (0.1-20 μM) inhibits topoisomerase II activity and induces site-selective DNA DSB in CCRF-CEM cells. Voreloxin (0.11, 0.33, 1, 3 μM) induces G2 arrest partially through topoisomerase II in A549 lung cancer cell line. Voreloxin cytotoxic activity requires DNA intercalation. However, Voreloxin (1-9 μM) does not generate significant levels of ROS[1]. Voreloxin has potent cytotoxic activity in AML cell lines MV4-11 and HL-60, with IC50s of 95 ± 8 nM and 884 ± 114 nM, respectively. Voreloxin in combination with cytarabine shows additive or synergistic activity in acute leukemia cell lines[2]. Voreloxin is active on the primary acute myeloid leukemia (AML) with a mean LD50 of 2.3 μM. The LD50 for voreloxin in myeloid cell lines NB4 and HL-60 is 0.59 μM ± 0.25 μM. Voreloxin causes accumulation of cells in the S and G2 phases of the cell cycle and acts on topoisomerase II[3]. Voreloxin (20 mg/kg, i.v.) alone results in 80% reduction in bone marrow cellularity of CD-1 mice by administration one dose every 4 days repeated twice (q4d ×2). voreloxin at 10 mg/kg in combination with cytarabine causes ablation of the marrow, dilation of sinusoids, and infiltration of adipocytes in mice. Voreloxin (20 mg/kg, i.v.) combined with cytarabine causes a reversible decrease in myeloid and lymphoid cells in bone marrow and peripheral blood CD-1 mice. voreloxin (10 mg/kg, q4d ×2) and cytarabine in combination causes reversible neutropenia with a more modest impact on platelets CD-1 mice[2]. Reference: |
 m.cnreagent.com
m.cnreagent.com