| CAS NO: | 106463-17-6 |
| 规格: | ≥98% |
| 包装 | 价格(元) |
| 25mg | 电议 |
| 50mg | 电议 |
| 100mg | 电议 |
| 250mg | 电议 |
| 500mg | 电议 |
| 1g | 电议 |
| Molecular Weight (MW) | 444.97 |
|---|---|
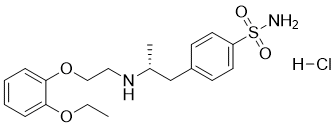
| Formula | C20H28N2O5S·HCl |
| CAS No. | 106463-17-6 (HCl); |
| Storage | -20℃ for 3 years in powder form |
| -80℃ for 2 years in solvent | |
| Solubility (In vitro) | DMSO: 88 mg/mL (197.8 mM) |
| Water: 17 mg/mL (38.2 mM) | |
| Ethanol: 8 mg/mL (18 mM) | |
| Solubility (In vivo) | O=S(C1=CC=C(C[C@H](NCCOC2=CC=CC=C2OCC)C)C=C1)(N)=O.[H]Cl |
| Synonyms | Tamsulosina hydrochloride, Tamsulosinum hydrochloride; Tamsulosin HCl; Pradif; Flomax; Omnic; |
| In Vitro | In vitro activity: Tamsulosin is a selective α1 receptor antagonist that has preferential selectivity for the α1A receptor in the prostate versus the α1B receptor in the blood vessels. Tamsulosin-treated patients had a 0.30-fold lower risk of developing acute urinary retention compared with control patients. None of the International Continence Society male questionnaire domain scores showed significant changes between the groups. tamsulosin can be recommended for treating men after catheterization for AUR, and can reduce the likelihood of the need for re-catheterization. Kinase Assay: Cell Assay: |
|---|---|
| In Vivo | Tamsulosin exhibits high plasma-protein binding, largely to α1-acid glycoprotein. It is metabolized, mainly by cytochrome P450 (CYP) 3A4 and CYP2D6 to compounds with low abundance, and 8.7–15% of an oral dose is excreted renally as the parent compound. The pharmacokinetics of tamsulosin are not affected to a major extent by age, and pharmacokinetic alterations in renally impaired patients relate largely to an increased concentration of α1-acid glycoprotein. Pharmacokinetic alterations with hepatic impairment are also only moderate, thus neither renal nor mild to moderate hepatic impairment necessitates dose adjustment. Early studies of tamsulosin IR in experimental animals, such as rats and dogs, showed rapid absorption of tamsulosin after oral administration (within 30–90 minutes) but absolute bioavailability of only 7–23% in rats and 30–42% in dogs. The absolute bioavailability of tamsulosin MR in fasted humans is approximately 100%. The tmax is typically about 5 hours (reported range of mean values 2.9-5.6 hours) in the fasted state and about 6 hours in the fed state (range 5.2-7.0 hours). Animal studies involving intravenous injection of radiolabelled tamsulosin and measurement of radiolabel in various tissues after 10 minutes have shown the presence of the drug in various tissues, ranked in the following order: kidney>lung≈heart>submaxillary gland>liver ≈spleen≈aorta≈vas deferens> prostate>>cerebral cortex, the latter being close to detection limits. Tamsulosin may not pass the blood-brain barrier. Tamsulosin undergoes extensive hepatic metabolism in rats and dogs, which excrete only 1.2% and 2.8%, respectively, in the urine as the parent compound. In humans, tamsulosin also undergoes extensive hepatic metabolism but apparently to a somewhat smaller degree than in rats or dogs, as 8.7–15% of an oral dose is excreted in the urine in a non-metabolized form. The speed of tamsulosin elimination from the body varies markedly between species, e.g. tamsulosin is eliminated faster in rats and dogs than in humans. |
| Animal model | Female Wistar rats |
| Formulation & Dosage | Dissolved in saline; 0.1 and 1 μg/kg; s.c. injection |
| References | Int J Urol. 2014 Feb;21(2):164-8; BJU Int. 2005 Feb;95(3):354-7. |
 m.cnreagent.com
m.cnreagent.com